


Idiopathic Pulmonary Fibrosis as a Challenge for the Polish Healthcare System
-
Copyright
© 2015 PRO MEDICINA Foundation, Published by PRO MEDICINA Foundation
User License
The journal provides published content under the terms of the Creative Commons 4.0 Attribution-International Non-Commercial Use (CC BY-NC 4.0) license.
Authors
| Name | Affiliation | |
|---|---|---|
Marcin Kwiecień |
Aestimo s.c., Kraków, Poland |
|
Monika Homa |
Aestimo s.c., Kraków, Poland |
|
Małgorzata Budasz-Świderska |
Roche Polska Sp. z o.o., Warszawa, Poland |
|
Marcin Kaczor |
Aestimo s.c., Kraków, Poland Uniwersytet Jegielloński Collegium Medicum, Kraków, Poland |
Idiopathic pulmonary fibrosis (IPF) is a rare disease characterised by age-dependent incidence, unclear aetiology and progressive course. Given the complexity of the diagnostic pathway and limited therapeutic options, and according to the fact that the Polish society is ageing, healthcare and social security expenditure are considerable and likely to rise. Until recently, there was no effective treatment for patients with IPF. After diagnosis only procedures that relieve the symptoms of the disease and treat its complications or lung transplantation were offered. In recent years a huge interest in this clinical entity has been seen. This resulted in the introduction of new drugs, whose efficacy has been confirmed in reliable clinical trials and whose slow the progression of the disease and improve survival of patients. The first such drug was pirfenidone; however such treatment is not available in Poland. Furthermore, due to the inadequacy of Polish legislative reimbursement solutions, which do not take into account the specific nature of rare disease, there are not used any favourable solution during the assessment of public funding of orphan drugs. This is contrary to European consensuses on an egalitarian approach to the use of orphan drugs. It is necessary to develop standards of care for IPF patients, including systemic solutions and the inclusion of the drugs used in the treatment of IPF on the reimbursement list. In long term this will reduce the financial burden associated with the disease, and thus reduce the direct and indirect costs resulting from its prevalence.
Introduction
Idiopathic pulmonary fibrosis (IPF) belongs to the group of diseases known as idiopathic interstitial pneumonia [1], and it is an example of a rare disease [2]. IPF is characterized by chronic, progressive, non-neoplastic and non-infectious process [1,3–4]. This leads to irreversible damage to the pulmonary parenchyma, entailing dramatic consequences such as considerable functional impairment and early disability of the affected individuals [5–6]. The precise pathomechanism underlying the disease is still not clear [4,7–8]. IPF is presumed to be a challenge to the healthcare system in Poland for multiple factors, both medical and economical [9–11]. Regarding lack of preventive measures and changes in the Polish demographic profile, early and accurate diagnosis as well as effective treatment should be one of the priorities in the healthcare system. Recent years have witnessed a genuine interest in this issue as well as major changes in the therapeutic approach, which has resulted in the introduction of new medicinal products [8,12], used in a number of other countries but still unavailable in Poland.
Disease characteristics
IPF is defined as a “specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults, limited to the lungs, and associated with a specific histopathological and/or radiological pattern of usual interstitial pneumonia (UIP)” [1], (International Classification of Diseases ICD-10 – J84.1).
Despite numerous studies into the pathogenic causes of the disease clear and coherent theory on its aetiology and pathogenesis is yet not provided [1,4,13]. The opinion that seems to be closest to the truth is that the mechanism underlying IPF is multifactorial [4,13]. As a result of a number of independent events, pathological activation of intracellular pathways occurs and the process of fibrotic transformation of pulmonary parenchyma is activated. One cannot but mention the fact that the processes are dependent on genetic predispositions and environmental factors [4].
Of the numerous risk factors for IPF, with regard to the Polish healthcare system burden, age, male sex, history of viral infection, type 2 diabetes mellitus and cigarette smoking are considered crucial [1]. This choice is due to the fact that the Polish society is ageing – according to the Central Statistical Office of Poland (GUS), in 2050 individuals aged over 65 will account for 32.7% of the general population, which means a two-fold increase relative to 2015 [14]. In addition, life expectancy will increase for both females and males [14].
The natural course of the disease is progressive and the prognosis – unfavourable [1,15,16]. In German studies involving a cohort of individuals selected from the register of IPF patients, mean age at diagnosis was about 68 years, and the diagnosis was typically established about four years after the onset of first symptoms [17]. The fact that the time needed to establish the diagnosis is so long may be related to non-characteristic symptoms of the disease, which means that other possible causes of interstitial lung diseases must be ruled out before the right diagnosis may be established [17–18]. In addition, the diagnosis needs to be established on the basis of such examinations as high-resolution computed tomography or lung biopsy with histopathological assessment [15,19]. Thus, it requires close collaboration between various specialists in an experienced team [19]. Given the above, it is clear that the risk of wrong diagnosis is high [19], which contributes to the delay in treatment initiation or to the initiation of treatment without clear indications, which in turn generates additional costs.
The most common signs and symptoms of IPF include dry cough and breathlessness, initially only on exertion and with time also at rest [1], which translates into a sudden reduction in the patient’s quality of life [20]. What is particularly important, in the majority of cases the progressive course of the disease leads to premature death – less than 15% of patients live longer than 10 years. The most common causes of death include: respiratory failure (40%), heart failure, ischaemic heart disease, infections, pulmonary embolism and lung cancer (4–15%) [21]. It should be noted that five-year survival rates for IPF are lower than for many malignant neoplasms (20–30%, with median survival ranging between 2.5 and 3.5 years) [22]. It should also be noted that in the course of the disease a number of comorbidities such as pulmonary hypertension and oesophageal reflux disease [1,19,23] develop, which is yet another argument for early diagnosis and prompt initiation of effective treatment.
Epidemiology
IPF is the most common diagnosis related to diseases classified as interstitial pneumonia, accounting for 47–64% of the diagnoses [24]. Recent years have brought about an increase in new diagnoses of IPF [25], probably as a result of the diagnostic process optimisation and improved life expectancy [26]. According to the Orphanet report, the incidence of IPF in the European Union countries, estimated on the basis of the systematic literature review, is 16.7:100,000 individuals [27]. A similar value has been given in the European Lung White Book [11]. Overall, the incidence and prevalence of IPF as reported in literature are lower in the European and Asian countries as compared with the USA [28]. This may result from differences in classifications and diagnostic criteria for IPF, which have changed over years, as well as varying methodologies of the studies [25,28]. Thus, the hypothesis that the epidemiological parameters given in literature may be of limited value for the precise estimation of the Polish population of IPF patients appears to be true. Precise depiction of the epidemiological situation in Poland may be difficult also because of the fact that no register of such patients is established. In most European countries more or less advanced registers of IPF cases are kept. Not so long ago, an initiative was announced to widen the Czech national register to include the Central and Eastern European countries. Thus, in 2014, the European MultiPartner IPF Registry (EMPIRE) was created. In addition to the Czech Republic, it covers pulmonary centres in Hungary, Serbia and Slovakia as well as six centres in Poland. As of now (December 2015), the total number of registered IPF cases is 681, including 96 Polish patients [29]. By comparison, in the INSIGHT–IPF register, which has been kept since November 2012 and includes leading pulmonary centres in Germany, the number of registered patients (as of 27 October 2015) is 502 [17]. Establishing both national and international registers of patients with IPF brings about definite advantages. In the future, they may become a basis for compiling precise characteristics of the disease in question. For the time being, given the present situation, drawing conclusions as to the epidemiology on the basis of these registers would entail the risk of bias.
IPF treatment options
Due to the lack of availability of the pirfenidone and nintedanib for Polish patients (drugs that slow the progression of the disease, prolong time to disease progression and improve survival of patients) current therapy of patients with IPF in Poland is based on the non-pharmacological management, which include lung transplantation, oxygen therapy or pulmonary rehabilitation [1]. Limited access to optimum treatment modalities, understood as effective causal treatment, has an additional negative effect on the prognosis in this disease.
Not so long ago, an immune mechanism was considered underlying the pathogenesis of spontaneous IPF. Therefore, international clinical guidelines of the American Thoracic Society/European Respiratory Society/ Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) (valid in 2000–2011) [15,30] recommended the use of immunosuppressants in this group of patients. The PANTHER-IPF study, in which triple therapy with N-acetylcysteine, prednisone and azathioprine was assessed, showed that it had no effect or even had negative effects such as more frequent hospital admissions and an increased risk of complications or death [31–33]. Likewise, there is no evidence for the effectiveness of cyclosporine [34], interferon-γ [35–36] or etanercept [37], therefore the use of immunosuppressants in patients with IPF is no longer recommended (Table 1.). Another treatment option which was recommended in older clinical guidelines for a long time (valid to 2011) was corticosteroids therapy. It is no longer recommended (Table 1.) as there is no evidence of its effectiveness [15,38–39], and the use of high dose corticosteroids is allowed only during disease exacerbation [15,26].
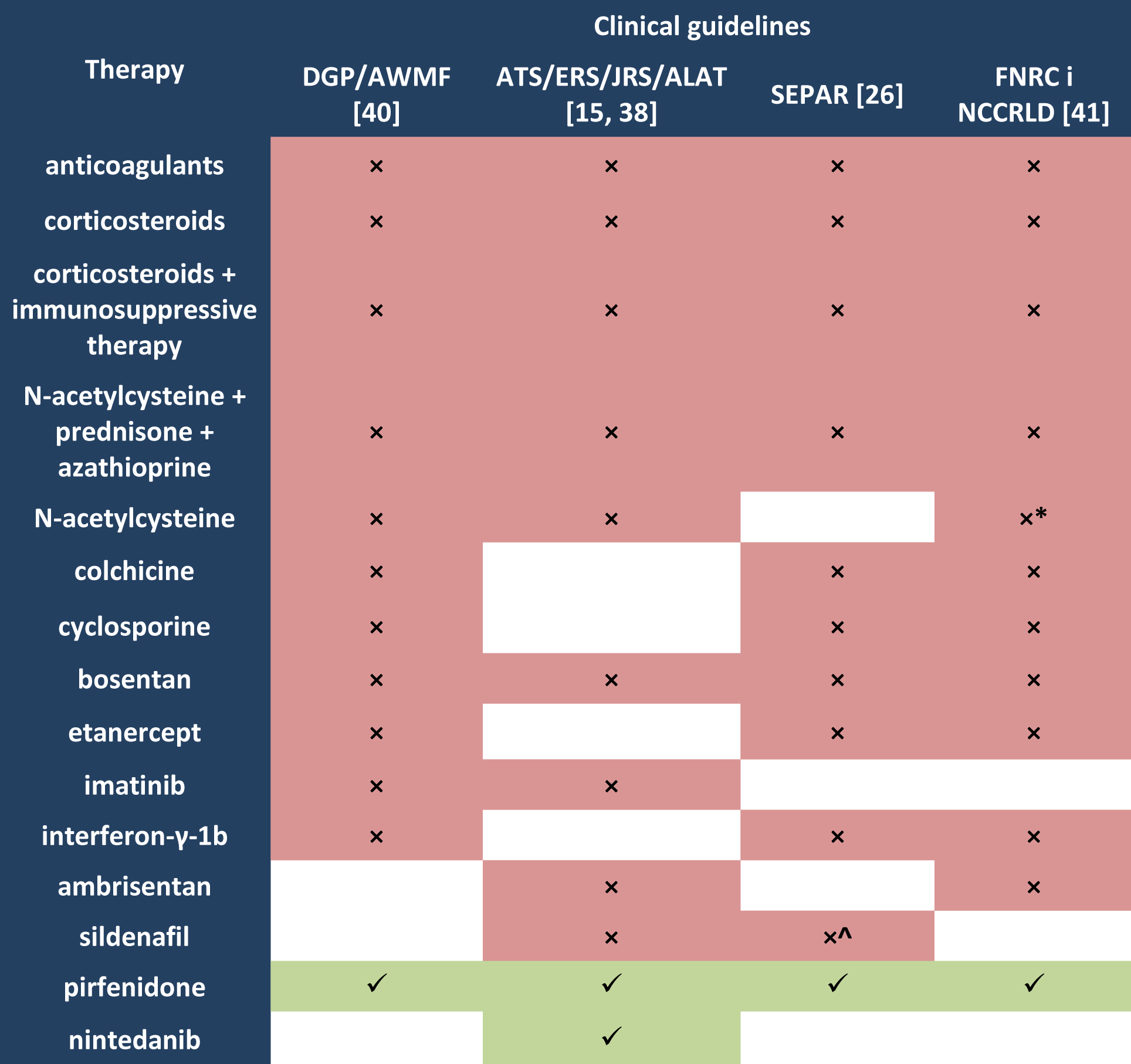
Table 1. Pharmacotherapy of idiopathic pulmonary fibrosis – clinical guidelines
Legend to Table 1.
ALAT Latin America Thoracic Association;
ATS American Thoracic Society;
AWMF Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften;
DGP Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin;
ERS European Respiratory Society;
FNRC French National Reference Centre;
JRS Japanese Respiratory Society;
NCCRLD Network of Competence Centres for Rare Lung Diseases;
SEPAR Spanish Society of Pneumology and Thoracic Surgery;
× not recommended;
√ recommended;
^ except patients with IPF and moderate-severe pulmonary hypertension [26];
* except some patients with a definite diagnosis of IPF, if treatment with an approved drug is not indicated
after having considered participation in a therapeutic clinical trial [41].
Pirfenidone and nintedanib
The first medicinal products approved for the treatment of IPF are Esbriet® and Ofev® [42]. They are the only available and recommended causal treatments (Table 1.). The active substance in these medicinal products has different mechanisms of action, leading to the inhibition of fibroblast proliferation. Pirfenidone (Esbriet®) has both anti-fibrotic and anti-inflammatory properties [8] – it reduces the production of proteins and cytokines involved in fibrotic transformation [43], while nintedanib (Ofev®) is a triple tyrosine kinase inhibitor [44].
The efficacy of pirfenidone was assessed in multicentre, randomised, placebo-controlled clinical trials: ASCEND [45], CAPACITY1 and CAPACITY2 [46], which involved a total of 1,334 Caucasian patients, and the SP2 [47] and SP3 [48] trials, which involved a total of 374 Asian patients. The ASCEND, CAPACITY1 and CAPACITY2 studies included patients with confirmed diagnosis of IPF, characterised by the FVC values at 50–90% of the predicted value and DLCO > 30–35% of the predicted value [45–46], which corresponded to mild to moderate disease, in accordance with the criteria adopted in IPF clinical trials (% FVC ≥ 50–55%, % DLCO ≥ 35–40%) [49]. The SP2 and SP3 trials included only patients with IPF whose severity was assessed in accordance with the Japanese guidelines [50], on the basis of partial pressure of oxygen in arterial blood (PaO2) and the lowest saturation (SpO2) during exercise. As the ASCEND trial has shown, therapy with pirfenidone significantly prolongs progression free survival , HR = 0.57 (95% CI: 0.43; 0.77), p < 0.0001 [45]. The study has also shown that the linear decrease in the FVC values was significantly lower in the pirfenidone group as compared with the placebo group (-164 ml vs -290 ml, p < 0.001) [45]. It is noteworthy that the outcomes of phase III studies of the pirfenidone effect on mortality rates were coherent. A pooled analysis of the ASCEND [45], CAPACITY1 and CAPACITY2 [46] trials has shown that therapy with pirfenidone significantly reduces the risk of death from any cause by 48% (HR = 0.52 [95% CI: 0.31; 0.87]; p = 0.01) and death from IPF by 68% (HR = 0.32 [95% CI: 0.14; 0.76]; p = 0.006) within one year [45]. The results are particularly significant when the natural course of the disease and unfavourable prognosis are taken into account.
, HR = 0.57 (95% CI: 0.43; 0.77), p < 0.0001 [45]. The study has also shown that the linear decrease in the FVC values was significantly lower in the pirfenidone group as compared with the placebo group (-164 ml vs -290 ml, p < 0.001) [45]. It is noteworthy that the outcomes of phase III studies of the pirfenidone effect on mortality rates were coherent. A pooled analysis of the ASCEND [45], CAPACITY1 and CAPACITY2 [46] trials has shown that therapy with pirfenidone significantly reduces the risk of death from any cause by 48% (HR = 0.52 [95% CI: 0.31; 0.87]; p = 0.01) and death from IPF by 68% (HR = 0.32 [95% CI: 0.14; 0.76]; p = 0.006) within one year [45]. The results are particularly significant when the natural course of the disease and unfavourable prognosis are taken into account.
The efficacy of nintedanib as compared with placebo was assessed in three randomised clinical trials: TOMORROW [51], INPULSIS-1 and INPULSIS-2 [52]. The trials were similar to the pirfenidone trials in terms of population (1,404 subjects) and disease severity (FVC ≥ 50% of the predicted value; DLCO 30–79% of the predicted value). In both the INPULSIS-1 and the INPULSIS-2 trials a significant effect of nintedanib on the subjects’ lung function was seen – the annual decrease in forced vital capacity (FVC) as compared with the placebo group was -114.7 ml vs -239.9 ml (p < 0.001) [51] and -113.6 ml vs -207.3 ml (p < 0.001), respectively [52]. The results show a reduced rate of IPF progression achieved with nintedanib. A pooled analysis of the INPULSIS-1 and INPULSIS-2 data did not reveal any effect of the drug on reduced all-cause mortality rates (HR = 0.70 [95% Cl: 0.43; 1.12]; p = 0.14) or respiratory mortality rates (HR = 0.74 [95% Cl: 0.41; 1.34]; p = 0.34) [52].
It is noteworthy that both products are similar in terms of the safety profile. During treatment with pirfenidone, as compared with the control group, the following adverse events were more common: nausea (in 36% vs 17% in CAPACITY1 and CAPACITY2, respectively [46],and 36.0% vs 13.4% in ASCEND [45]) and rash (32% vs 12% in CAPACITY1 and CAPACITY2, respectively [46], and 28.1% vs 8.7% in ASCEND [45]). During treatment with nintedanib, as compared with placebo, the following adverse events were more common: diarrhoea (in 61.5% vs 18.6% and 63.2% vs 18.3% in INPULSIS-1 and INPULSIS-2, respectively), nausea (22.7% vs 5.9% and 26.1% vs 7.3%) and vomiting (12.9% vs 2.0% and 10.3% vs 3.2%) [52].
Current clinical guidelines do not indicate which of the two products should be used first [38]. So far, only one phase II clinical trial was aimed at the assessment of safety and pharmacokinetics of both products used either in monotherapy or in combination [53]. However, it should be noted that the trial had a number of limitations. Pirfenidone as monotherapy was received only by 10% of subjects (5/50) as compared with 34% (17/50) of subjects receiving nintedanib as monotherapy. In addition, the trial involved only Japanese subjects [53], which makes it difficult to compare the products and relate the data obtained to the European population.
Disease burden
As has already been mentioned, IPF poses a substantial burden on the healthcare system. Precise estimation of costs related to the diagnosis of IPF is difficult due to the factors detailed above – not fully known etiopathogenesis, difficulty in diagnostic evaluation and classification and uncertainty over the population estimation.
Potential costs related to interstitial lung diseases, as listed in the European Lung White Book [11], include premature mortality, particularly in the male population. As a result of the disease, work productivity decreases and some patients have to resign from professional career. Furthermore, they require costly supportive management (pulmonary rehabilitation, oxygen therapy). Exacerbations generate additional healthcare costs such as anti-inflammatory treatment or antibiotic therapy. Poland belongs to the group of countries with the highest age-standardised rates of hospital admissions in the course of ILD (> 40:100,000 of inhabitants) [11].
The Łazarski University from Warsaw report concerns the current healthcare burden related to interstitial lung diseases, including IPF. The number of IPF patients treated in the in-patient setting in 2014 exceeded 4,200, which means an increase relative to the previous year, and estimated financial costs of these hospital stays exceeded PLN 15 million. A median hospital stay lasted seven days, and hospital treatment concerned mostly individuals aged over 61. The Łazarski University experts have estimated that annual healthcare costs for IPF are higher as compared with other diseases. Patients with interstitial diseases, including IPF, pose a considerable social burden, particularly in terms of benefits paid by the Social Insurance Institution (ZUS). In 2014, the total number of sick leave days related to the diagnosis code J84 was 97.4 thousand days. According to the data presented in the Łazarski University report, the total indirect costs incurred by ZUS in 2013 in relation to another interstitial pulmonary disease exceeded PLN 18 million for pension, sickness and rehabilitation benefits [54].
The weakness of the data presented in the report lies in the fact that the ICD-10 code J.84 includes also other diseases. In addition, the values of social insurance benefits concern only the ZUS-insured individuals and one may assume that they are underestimated. Nevertheless, the data are to give a general idea of the financial costs related to the prevalence of spontaneous pulmonary fibrosis and, by comparison with the entire document, indicate that measures should be taken to improve the situation of Polish patients diagnosed with this disease. Only optimisation of healthcare and reimbursement of therapies which have been successfully used in Europe may prevent the increase in healthcare expenditure related to IPF and reduce the socio-economic losses seen in the natural course of the disease [54].
Mortality reduction may be achieved by earlier implementation of specialist care, because delayed initiation of treatment in a reference centre, caused by prolonged diagnostic evaluation or its unavailability, was associated with higher mortality rates [55]. The overall treatment costs may be reduced mainly by using clinically effective therapies such as pirfenidone and nintedanib [56].
Having analysed reimbursement recommendations produced by leading foreign HTA agencies, one should note that the need has already been appreciated. All the agencies which assessed Esbriet® issued positive reimbursement recommendations. Similarly, in the case of Ofev® most decisions were positive (except for the Australian agency) (Table 2.).
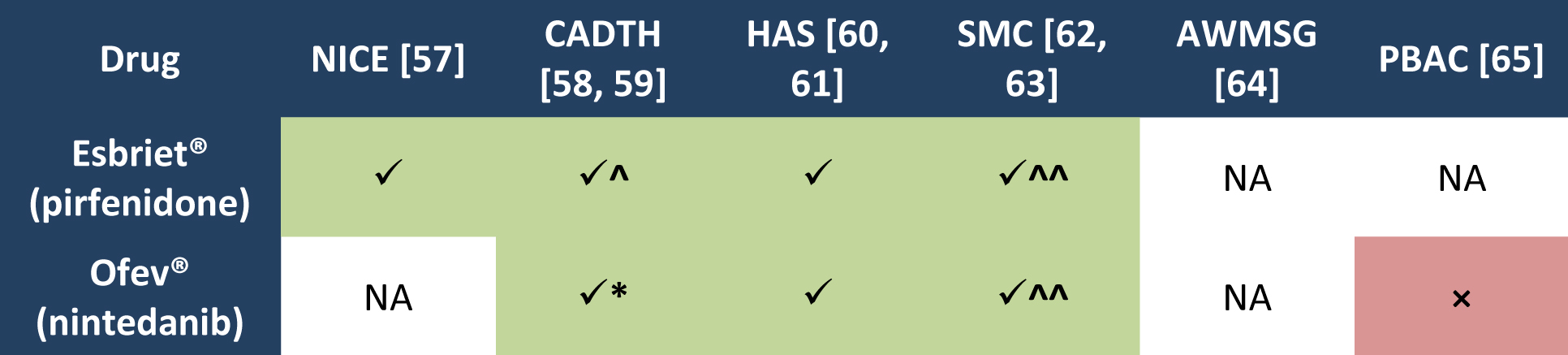
Table 2. Recommendation for reimbursement of Esbriet® and Ofev®
Legend to Table 2.
AWMSG All Wales Medicines Strategy Group;
CADTH Canadian Agency for Drugs and Technologies in Health
HAS Haute Autorité de Sainté
NICE National Institute for Health and Clinical Excellence
PBAC Pharmaceutical Benefits Advisory Committee
SMC Scottish Medicines Consortium
NA not applicable
× negative recommendation
√ positive recommendation;
^ pirfenidone may be use to treatment of IPF when following criteria and conditions are met: mild to moderate IPF, defined as forced vital capacity (FVC) greater than or equal to 50% of predicted; stable disease, defined as FVC not decreased by ≥ 10% during the previous 12 months; treatment discontinued if FVC declines by ≥ 10% within any 12-month period while receiving therapy; patient is under the care of a specialist with experience in the diagnosis and management of patients with IPF; substantial price reduction [58].
* nintedanib may be use to treatment of IPF when following criteria and conditions are met: forced vital capacity (FVC) greater than or equal to 50% of predicted; treatment with nintedanib should be discontinued if absolute FVC declines by ≥ 10% within any 12-month period while receiving therapy. Conditions: under the care of a specialist with experience in the diagnosis and management of IPF; drug plan cost for nintedanib must not exceed the drug plan cost for pirfenidone [59].
^^ for use in patient with a predicted forced vital capacity (FVC) less than or equal to 80% [62,63].
It should be emphasized that most of the HTA agency took into account during refund process two aspects: the specificity of rare diseases, which is the IPF and the lack of effective therapeutic alternatives. As for the real reimbursement costs incurred by the European countries which belong to the European Free Trade Association (EFTA), it should be noted that in most of them pirfenidone is now available free of charge. Furthermore, ranking EFTA member states in order of growing GDP shows that nintedanib is reimbursed only in the countries with higher GDP (while pirfenidone is available free of charge in all of them), and pirfenidone is reimbursed in countries with GDP similar to that of Poland (Figure 1.).
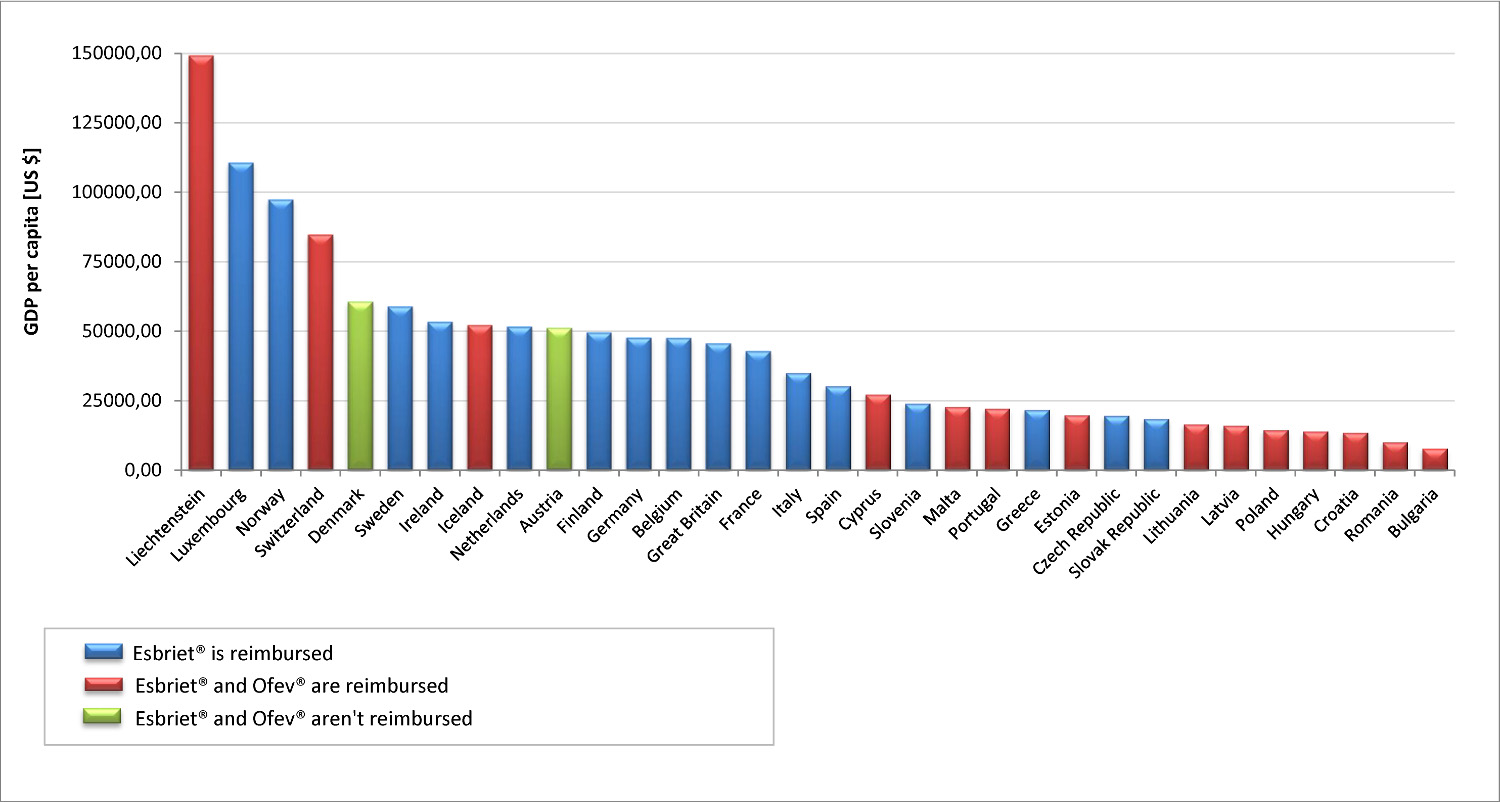
Figure 1. Reimbursement of Esbriet® and Ofev® in EFTA countries
Legend to Figure 1.
Reimbursement status of Esbriet® and Ofev® according to data available at [66-97] and [98]. GDP per capita for the year 2015, or in the absence of data for the next year with available data according to [99].
As regards treatment options now available to Polish patients with IPF, in particular the non-reimbursement of new technologies – pirfenidone and nintedanib, one should note that the situation is inconsistent with the joint position adopted by the parliamentary groups on rare diseases and on oncology, which reflected an egalitarian approach to funding orphan drugs based on conclusions reached in the EUROPLAN November 2013 debate [100]. The issues, in particular the Polish situation, have been discussed in more detail in the document entitled: “Systemic assumptions for the development of the National Rare Diseases Plan for 2013–2017” [101]. Authors of this paper point to the fact that the specific nature of this group of diseases has not been taken into consideration in the health technology assessment procedure. Since the scientific data are scarce, the reports on orphan drugs efficacy are questioned, which – through the negative decision of the Agency for Health Technology Assessment and Tariff System (AOTMiT) – contributes to the fact that patients are denied access to therapeutic regimens widely used in European countries. It is noteworthy than in a number of EU countries the restriction related to the rare occurrence of diseases such as IPF is taken into consideration in the reimbursement processes [101].
Summary
In the view of situation of patients with IPF analysed in this study, it should be noted that both unknown aetiology and the difficulties related to the diagnosis of the disease result in a very unfavourable prognosis and also therapies currently used in Poland do not allow for inhibiting the progression of IPF. The severity of symptoms and the number of comorbidities occurring in the natural course of this condition has a dramatic impact on the deterioration of the quality of life of patients, as time goes by excluding them from active participation in social life and leading to death. This situation is associated with high direct and indirect costs associated with IPF, which translates into considerable expenses incurred by both the health system and social security system.
In view of the lack of access in Poland for effective causal treatment of the disease, it should be also noted that the IPF remains an unmet medical need. These condition belongs to the group of rare diseases which, given the current inadequacy of Polish legislative solutions to the specific nature of these diseases significantly delays the reimbursement of orphan drugs. Therefore Polish patients do not receive treatment recommended in clinical guidelines, which is already available for patients from other European countries, including those with GDP comparable to that of the Polish GDP. From these point of view amendments to the reimbursement law appear to be necessary. First, however, pirfenidone and nintedanib should be made available as soon as possible, in order to meet the challenge that IPF presents to the Polish healthcare system.
- Rowińska-Zakrzewska E, Bestry I. Idiopatyczne włóknienie płuc. Interna Szczeklika. Gajewski P. (ed.), Medycyna Praktyczna, Krakow 2015;729–734
- Orphanet Report Series. Rare disease collection. List of rare diseases and synonyms: Listed in alphabetical order. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/List_of_rare_diseases_in_alphabetical_order.pdf
- Demkow U. Immunopathogenesis of idiopathic pulmonary fibrosis. Pneumonol Alergol Pol. 2014; 82(1):55–60; doi: 10.5603/PiAP.2014.0009
- Wolters PJ, Collard HR, Jones KD. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu Rev Pathol. 2014; 9:157–179; doi: 10.1146/annurev-pathol-012513-104706
- Belkin A, Swigris JJ. Health-related quality of life in idiopathic pulmonary fibrosis: where are we now? Curr Opin Pulm Med. 2013; 19(5):474–9; doi: 10.1097/MCP.0b013e328363f479
- Swigris JJ, Kuschner WG, Jacobs SS, Wilson SR, Gould MK. Health-related quality of life in patients with idiopathic pulmonary fibrosis: a systematic review. Thorax. 2005; 60(7):588–94; doi:10.1136/thx.2004.035220
- Bellaye PS, Kolb M. Why do patients get idiopathic pulmonary fibrosis? Current concepts in the pathogenesis of pulmonary fibrosis. BMC Med. 2015; 13:176; doi: 10.1186/s12916-015-0412-6
- Tzouvelekis A, Bonella F, Spagnolo P. Update on therapeutic management of idiopathic pulmonary fibrosis. Ther Clin Risk Manag. 2015; 11:359–70; doi: 10.2147/TCRM.S69716. eCollection 2015
- Lee AS, Mira-Avendano I, Ryu JH, Daniels CE. The burden of idiopathic pulmonary fibrosis: an unmet public health need. Respir Med. 2014; 108(7):955–67; doi: 10.1016/j.rmed.2014.03.015
- Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ. 2012; 15(5):829–35; doi: 10.3111/13696998.2012.680553
- European Respiratory Society. European Lung White Book. Part C. Chapter 22. Interstitial lung disease. Available from: http://www.erswhitebook.org/chapters/interstitial-lung-diseases/
- Sergew A, Brown KK. Advances in the treatment of idiopathic pulmonary fibrosis. Expert Opin Emerg Drugs. 2015; 20(4):1–17; doi: 10.1517/14728214.2015.1102886
- Spagnolo P, Rossi G, Cavazza A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev. Clin. Immunol. 2014; 10(8):1005–1017; doi:10.1586/1744666X.2014.91705
- Demographic profile of the elderly and consequences of the Polish population ageing in light of the forecast for 2014–2050. Available from: http://stat.gov.pl/obszary-tematyczne/ludnosc/ludnosc/sytuacja-demograficzna-osob-starszych-i-konsekwencje-starzenia-sie-ludnosci-polski-w-swietle-prognozy-na-lata-2014-2050,18,1.html
- Raghu G, Collard HR, Egan JJ, Martinez FJ, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011; 183(6):788–824; doi:10.1164/rccm.2009-040GL
- Elmufdi F, Henke CA, Perlman DM, Tomic R, Kim HJ. Novel mechanisms and treatment of idiopathic pulmonary fibrosis. Discov Med. 2015 Sep; 20(109):145–53
- Behr J, Kreuter M, Hoeper MM et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J. 2015; 46(1):186–96; doi: 10.1183/09031936.00217614
- Valeyre D. Towards a better diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev. 2011; 20(120):108–13; doi: 10.1183/09059180.00001611
- Spagnolo P, Tonelli R, Cocconcelli E, Stefani A, Richeldi L. Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges. Multidiscip Respir Med. 2012; 7(1):42; doi: 10.1186/2049-6958-7-42
- Ryerson CJ, Donesky D, Pantilat SZ, Collard HR. Dyspnea in idiopathic pulmonary fibrosis: a systematic review. J Pain Symptom Manage. 2012; 43(4):771–82; doi: 10.1016/j.jpainsymman.2011.04.026
- Ziora D. Samoistne śródmiąższowe zapalenia płuc i zapalenia oskrzelików. Wielka Interna. Pulmonologia. Część II. Wyd. 1. Antczak A (ed.). Medical Tribune Polska, Warsaw 2010; 317–321
- Ley B, Harold R. Collard HR, King TE Jr. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis Am J Respir Crit Care Med. 2011; 183(4):431–40; doi: 10.1164/rccm.201006-0894CI
- Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med. 2015; 109(6):661–70; doi: 10.1016/j.rmed.2015.02.002
- Papla B. Śródmiąższowe idiopatyczne zapalenie płuc. Pol J Pathol 2010; 1(1):1323
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015; 46(3):795–806; doi: 10.1183/09031936.00185114
- Xaubet A, Ancochea J, Bollo E, Fernández-Fabrellas E, Franquet T, Molina-Molina M et al. A. Normativa sobre el diagnóstico y tratamiento de la fibrosis pulmonar idiopática. Arch. Bronconeumol. 2013; 49(8):343–353; doi:10.1016/j.arbres.2013.03.011
- Orphanet Report Series. Rare Diseases collection. Prevalence and incidence of rare diseases: Bibliographic data. Prevalence, incidence or number of published cases listed by diseases (in alphabetical order). Number 1. July 2015. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
- Caminati A, Madotto F, Cesana G, Conti S, Harari S. Epidemiological studies in idiopathic pulmonary fibrosis: pitfalls in methodologies and data interpretation. Eur Respir Rev. 2015; 24(137):436–44; doi: 10.1183/16000617.0040-2015
- EMPIRE (European MultiPartner IPF Registry). Available from: http://empire.registry.cz/
- American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000; 161(2 Pt 1):646–64
- Behr J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012; 367(9):869; author reply 870–871; doi:10.1056/NEJMc1207471#SA1
- Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366:1968–1977; doi:10.1056/NEJMoa1113354
- Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014; 370(22):2093–2101; doi:10.1056/NEJMoa1401739
- Homma S, Sakamoto S, Kawabata M et al. Cyclosporin treatment in steroid-resistant and acutely exacerbated interstitial pneumonia. Intern Med. 2005 Nov; 44(11):1144–50
- Raghu G, Brown KK, Bradford WZ et. al. Idiopathic Pulmonary Fibrosis Study Group. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2004; 350(2):125–133; doi:10.1056/NEJMoa030511
- King TE, Albera C, Bradford WZ et. al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 2009; 374(9685):222–228; doi:10.1016/S0140-6736(09)60551–1
- Raghu G, Brown KK, Costabel U et al.Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med 2008; 178:948–955; doi: 10.1164/rccm.200709-1446OC
- Raghu G, Rochwerg B, Zhang B et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guide-line: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. American Thoracic Society Documents. Am J Respir Crit Care Med. 2015 Jul 15; 192(2):e3–19; doi: 10.1164/rccm.201506-1063ST
- Richeldi L. Clinical trials of investigational agents for IPF: a review of a Cochrane report. Respir Res. 2013; 14(Suppl 1):S4; doi: 10.1186/1465-9921-14-S1-S4
- Behr J, Günther A, Ammenwerth W et al. S2K-Leitlinie zur Diagnostik und Therapie der idiopathischen Lungenfibrose. Pneumologie 2013; 67(02):81–111; doi:10.1055/s-0032-1326009
- Cottin V, Crestani B, Valeyre D et al. Diagnosis and management of idiopathic pulmonary fibrosis: French practical guidelines. Eur. Respir. Rev. 2014; 23(132):193–214; doi:10.1183/09059180.00001814
- Myllärniemi M, Kaarteenaho R. Pharmacological treatment of idiopathic pulmonary fibrosis – preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. Eur Clin Respir J. 2015; 2. doi: 10.3402/ecrj.v2.26385
- Summary of Product Characteristics for Esbriet®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002154/human_med_001417.jsp&mid=WC0b01ac058001d124
- Summary of Product Characteristics Ofev®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003821/human_med_001834.jsp&mid=WC0b01ac058001d124
- King Jr TE, Bradford WZ, Castro-Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New Engl J Med. 2014; 370(22):2083–2092; doi: 10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet. 2011; 377(9779):1760–1769; doi: 10.1016/S0140-6736(11)60405–4
- Azuma A, Nukiwa T, Tsuboi E et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005; 171(9):1040–1047
- Taniguchi H, Ebina M, Kondoh Y et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010; 35(4):821–82; doi: 10.1183/09031936.00005209
- Kolb M, Collard HR. Staging of idiopathic pulmonary fibrosis: past, present and future. Eur. Respir. Rev. 2014; 23(132):220–224. doi:10.1183/09059180.00002114
- Homma S, Sugino K, Sakamoto S. The usefulness of a disease severity staging classification system for IPF in Japan: 20 years of experience from empirical evidence to randomized control trial enrollment. Respir. Investig. 2015; 53(1):7–12; doi:10.1016/j.resinv.2014.08.003
- Richeldi L, Costabel U, Selman M et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011; 365(12):1079–87; doi: 10.1056/NEJMoa1103690
- Richeldi L, du Bois RM, Raghu G et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014 May 29; 370(22):2071–82; doi: 10.1056/NEJMoa1402584
- Ogura T, Taniguchi H, Azuma A et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015; 45(5):1382–92; doi: 10.1183/09031936.00198013
- Biała Księga. Samoistne włóknienie płuc – aspekty kliniczne, ekonomiczne i systemowe ze szczególnym uwzględnieniem rekomendacji dotyczących optymalnego zarządzania chorobą. Gałązka-Sobotka M, Gierczyński J (ed.). Łazarski University, Warsaw, 2015
- Lamas DJ, Kawut SM, Bagiella E et al. Delayed Access and Survival in Idiopathic Pulmonary Fibrosis; Am J Respir Crit Care Med. 2011; 184(7): 842–847; doi: 10.1164/rccm.201104-0668OC
- Loveman E, Copley VR, Colquitt J et al. The clinical effectiveness and cost-effectiveness of treatments for idiopathic pulmonary fibrosis: a systematic review and economic evaluation. Health Technol Assess. 2015; 19(20):i–xxiv, 1–336; doi: 10.3310/hta19200
- National Institute for Health and Care Excellence. Available from: https://www.nice.org.uk/guidance/ta282
- Canadian Agency for Drugs and Technologies in Health. Pirfenidone. Available from: https://www.cadth.ca/pirfenidone-12
- Canadian Agency for Drugs and Technologies in Health. Nintedanib. Available from: https://www.cadth.ca/nintedanib
- Haute Autorité de Santé. Pirfenidone. Available from: http://www.has-sante.fr/portail/jcms/c_1238454/en/esbriet?xtmc=&xtcr=2
- Haute Autorité de Santé. Nintedanib. Available from: http://www.has-sante.fr/portail/jcms/c_2038122/en/ofev-nintedanib-inhibiteur-des-tyrosines-kinases?xtmc=&xtcr=3
- Scottish Medicines Consortium. Pirfenidone. Available from: https://www.scottishmedicines.org.uk/SMC_Advice/Advice/835_13_pirfenidone_Esbriet/pirfenidone_Esbriet
- Scottish Medicines Consortium. Nintedanib. Available from: https://www.scottishmedicines.org.uk/SMC_Advice/Advice/1076_15_nintedanib_Ofev/nintedanib_Ofev
- All Wales Medicines Strategy Group. Available from: http://www.awmsg.org/
- Pharmaceutical Benefits Scheme. Nintedanib. Available from: http://www.pbs.gov.au/info/industry/listing/elements/pbac-meetings/psd/2015-03/nintedanib-caps-ofev-psd1-2015-03
- Principality of Lichtenstein. Ministry for Social Affairs. Available from: http://www.regierung.li/ministries/ministry-for-social-affairs/links/
- Grand-Duché de Luxembourg. Ministre De La Santé. Available from: http://www.ms.public.lu/fr/index.php
- Ministry of Health and Care Services Norway. Available from: https://www.regjeringen.no/en/dep/hod/id421/
- Federal Office of Public Health Switzerland. Available from: http://www.bag.admin.ch/index.html?lang=en
- Denmark Ministry of Health. Available from: http://www.sum.dk/English.aspx
- Sweden. Ministry of Health and Social Affairs. Available from: http://www.government.se/government-of-sweden/ministry-of-health-and-social-affairs/
- Ireland. Department of Health. Available from: http://health.gov.ie/
- Iceland. Ministry of Welfare. Available from: http://eng.velferdarraduneyti.is/
- Netherlands. Ministry of Health, Welfare and Sport. Available from: https://www.government.nl/ministries/ministry-of-health-welfare-and-sport
- Austria. Federal Ministry of Health. Available from: http://bmg.gv.at/home/EN/Home
- Finland. Ministry of Social Affairs and Health. Available from: http://stm.fi/en/frontpage
- Germany. Federal Ministry of Health. Available from: http://www.bmg.bund.de/en.html
- Belgium. Federal Public Service. Health, Food Chain Safety and Environment. Available from: http://www.health.belgium.be/eportal?fodnlang=en
- Great Britain. Department of Health. Available from: https://www.gov.uk/government/organisations/department-of-health
- France. Ministère des Affaires sociales, de la Santé et des Droits des femmes. Available from: http://www.sante.gouv.fr/
- Italy. Ministerio della Salute. Available from: http://www.salute.gov.it/
- Spain. Ministerio de Sanidad, Servicios Sociales e Igualdad. Available from: http://www.msssi.gob.es/en/home.htm
- Cyprus. Ministry of Health. Available from: http://www.moh.gov.cy/moh/moh.nsf/index_en/index_en
- Republic of Slovenia. Ministry of Health. Available from: http://www.mz.gov.si/en/
- Malta. Ministry of Health. Available from: https://health.gov.mt/en/Pages/health.aspx
- Portugal. Ministério da Saúde. Available from: http://www.portaldasaude.pt/portal
- Greece. Ministry of Health. Available from: http://www.moh.gov.gr/
- Estonia. Sotsiaalministeerium. Available from: http://www.sm.ee/et
- Czech Republic Ministry of Health. Available from: http://www.mzcr.cz/
- Ministerstvo zdravotníctva Slovenskej republiky. Available from: www.health.gov.sk/
- Ministry of Health of The Republic of Lithuania. Available from: sam.lrv.lt/
- Latvia Ministry of Health. Available from: www.vm.gov.lv/en/
- Poland. Ministry of Health. Available from: http://www.mz.gov.pl/
- Hungary. Ministry of Health. Available from: www.eum.hu/english
- Ministry of Health of the Republic of Croatia. Available from: www.zdravlje.hr/en/ministry
- Romania. Ministerul Sanatatii. Available from: http://www.ms.ro/
- Bulgaria. Ministry of Health. Available from: www.mh.government.bg
- Application for reimbursement of Ofev and setting its official price. Verification Analysis. AOTMiT. Warsaw 2015. Available from: http://www.aotm.gov.pl/bip/index.php/zlecenia-mz-2015/829-materialy-2015/4123-107-2015-zlc
- The World Bank. GDP per capita. Available from: http://data.worldbank.org/indicator/NY.GDP.PCAP.CD
- Position of the Parliamentary Group on Rare Diseases and the Parliamentary Group on Oncology on the criterion of the threshold for the cost of gaining an additional quality-adjusted life year and applying the results of classic economic analysis of the profitability threshold – 9 September 2014. Available from: http://www.rzadkiechoroby.pl/materialy/Stanowisko%20Zespolow%20Parlamentarnych%20QUALY.pdf
- Systemic assumptions in the development of the NATIONAL RARE DISEASES PLAN for 2013–2017. Available from: http://www.rzadkiechoroby.pl/np/

